Matthias Geilhufe
About me
Group Theory Book

A course on group theory in solid state physics and photonics together with a detailed description about the application of our Mathematica group theory package GTPack can be found in our book Group Theory in Solid State Physics and Photonics: Problem Solving with Mathematica. The book contains hands-on examples covering basic group theory to advanced applications.
W. Hergert, M. Geilhufe, Group Theory in Solid State Physics and Photonics: Problem Solving with Mathematica, Wiley-VCH, ISBN: 978-3-527-41133-7, 2018
More information about the book and the Mathematica group theory package GTPack can be found at https://gtpack.org
Research
Materials informatics and Organic Materials Database
We develop the Organic Materials Database - OMDB, transfering methodology from data science into functional materials research. The OMDB hosts, electronic, crystal, and magnetic structure information of more than 40,000 synthesized organic molecular crystals and metal organic frameworks. The data is accumulated through high throughput calculations. The OMDB is free. It hosts advanced pattern matching functionality and machine learning based property prediction tools.
The OMDB can be accessed via https://omdb.mathub.io.
Selected publications:
- R. M. Geilhufe, B. Olsthoorn, A. V. Balatsky, Shifting computational boundaries for complex organic materials, Nature Physics (2021)
- S. S. Borysov, R. M. Geilhufe, and A. V. Balatsky, Organic materials database: An open-access online database for data mining, PLOS ONE, 12, 2, e0171501 (2017)
Covered by media:
- Using AI guides to find new materials for electronics and more (also available in Swedish)
- New data mining resource for organic materials available (Science Daily)
Symmetry Principles of Quantum Matter
Quantum matter describes a class of phenomena where quantum effects remain dominant over a wide range of energy and length scales. The realization of quantum matter is typically strongly bound to symmetry, symmetry breaking, and topology. I apply and develop group theory based methodology to investigate quantum matter in the static and time domain, with particular focus on Dirac materials and superconductors.
Selected publications:
- R. M. Geilhufe and W. Hergert, GTPack: A Mathematica Group Theory Package for Application in Solid-State Physics and Photonics, Frontiers in Physics, 6, 86 (2018)
- R. M. Geilhufe and A. V. Balatsky, Symmetry analysis of odd- and even-frequency superconducting gap symmetries for time-reversal symmetric interactions, Physical Review B, 97, 024507 (2018)
Dirac Matter
Dirac matter describes phases of matter with elementary excitations behaving as Dirac fermions. I am interested in the realization and protection of Dirac states. Furthermore, I investigate applications of Dirac matter as well as interaction effects.
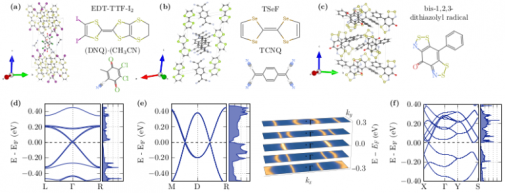
Selected publications:
- R. M. Geilhufe and B. Olsthoorn, Identification of strongly interacting organic semimetals, Physical Review B, 102, 205134 (2020)
- R. M. Geilhufe, F. Kahlhoefer, and M. W. Winkler, Dirac materials for sub-MeV dark matter detection: New targets and improved formalism, Physical Review D 101, 05505 (2020)
Full list of publications
Full list of publications can be found at google scholar.
Publications
A selection from Stockholm University publication database
-
Identification of strongly interacting organic semimetals
2020. R. Matthias Geilhufe, Bart Olsthoorn. Physical Review B 102 (20)
ArticleRead more about Identification of strongly interacting organic semimetalsDirac and Weyl point- and line-node semimetals are characterized by a zero band gap with simultaneously vanishing density of states. Given a sufficient interaction strength, such materials can undergo an interaction instability, e.g., into an excitonic insulator phase. Due to generically flatbands, organic crystals represent a promising materials class in this regard. We combine machine learning, density functional theory, and effective models to identify specific example materials. Without taking into account the effect of many-body interactions, we found the organic charge transfer salts [bis(3,4-diiodo-3',4'-ethyleneditio-tetrathiafulvalene), 2,3-dichloro-5,6-dicyanobenzoquinone, acetenitrile] [(EDT-TTF-I-2)(2)](DDQ)center dot(CH3CN) and 2, 2', 5, 5'-tetraselenafulvalene-7, 7, 8, 8-tetracyano-p-quinodimethane (TSeF-TCNQ) and a bis-1,2,3-dithiazolyl radical conductor to exhibit a semimetallic phase in our ab initio calculations. Adding the effect of strong particle-hole interactions for (EDT-TTF-I-2)(2)(DDQ)center dot(CH3CN) and TSeF-TCNQ opens an excitonic gap on the order of 60 and 100 meV, which is in good agreement with previous experiments on these materials.
-
Dirac materials for sub-MeV dark matter detection
2020. R. Matthias Geilhufe, Felix Kahlhoefer, Martin Wolfgang Winkler. Physical Review D 101 (5)
ArticleRead more about Dirac materials for sub-MeV dark matter detectionBecause of their tiny band gaps Dirac materials promise to improve the sensitivity for dark matter particles in the sub-MeV mass range by many orders of magnitude. We study several candidate materials and calculate the expected rates for dark matter scattering via light and heavy dark photons as well as for dark photon absorption. A particular emphasis is placed on how to distinguish a dark matter signal from background by searching for the characteristic daily modulation of the signal, which arises from the directional sensitivity of anisotropic materials in combination with the rotation of Earth. We revisit and improve previous calculations and propose two new candidate Dirac materials: bis(naphthoquinone)tetrathiafulvalene (BNQ-TTF) and Yb3PbO. We perform detailed calculations of the band structures of these materials and of ZrTe5 based on density functional theory and determine the band gap, the Fermi velocities, and the dielectric tensor. We show that in both ZrTe5 and BNQ-TTF the amplitude of the daily modulation can be larger than 10% of the total rate, allowing us to probe the preferred regions of parameter space even in the presence of sizable backgrounds. BNQ-TTF is found to be particularly sensitive to small dark matter masses (below 100 keV for scattering and below 50 meV for absorption), while Yb3PbO performs best for heavier particles.
-
Dynamically induced magnetism in KTaO3
2021. R. Matthias Geilhufe (et al.). Physical Review Research 3 (2)
ArticleRead more about Dynamically induced magnetism in KTaO3Dynamical multiferroicity features entangled dynamic orders: fluctuating electric dipoles induce magnetization. Hence, the material with paraelectric fluctuations can develop magnetic signatures if dynamically driven. We identify the paraelectric KTaO3 (KTO) as a prime candidate for the observation of the dynamical multiferroicity. We show that when a KTO sample is exposed to a circularly polarized laser pulse, the dynamically induced ionic magnetic moments are of the order of 5% of the nuclear magneton per unit cell. We determine the phonon spectrum using ab initio methods, and we identify T-1u as relevant phonon modes that couple to the external field and induce magnetic polarization. We also predict a corresponding electron effect for the dynamically induced magnetic moment, which is enhanced by several orders of magnitude due to the significant mass difference between electron and ionic nucleus.
-
Magnetic and Electronic Properties of Complex Oxides from First-Principles
2020. Martin Hoffmann (et al.). Physica status solidi. B, Basic research 257 (7)
ArticleRead more about Magnetic and Electronic Properties of Complex Oxides from First-PrinciplesThe theoretical treatment of complex oxide structures requires a combination of efficient methods to calculate structural, electronic, and magnetic properties, due to special challenges such as strong correlations and disorder. In terms of a multicode approach, this study combines various complementary first-principles methods based on density functional theory to exploit their specific strengths. Pseudopotential methods, known for giving reliable forces and total energies, are used for structural optimization. The optimized structure serves as input for the Green's function and linear muffin-tin orbital methods. Those methods are powerful for the calculation of magnetic ground states and spectroscopic properties. Within the multicode approach, disorder is investigated by means of the coherent potential approximation within a Green's function method or by construction of special quasirandom structures in the framework of the pseudopotential methods. Magnetic ground states and phase transitions are studied using an effective Heisenberg model treated in terms of a Monte Carlo method, where the magnetic exchange parameters are calculated from first-principles. The performance of the multicode approach is demonstrated with different examples, including defect formation, strained films, and surface properties.
-
Spin wave excitations of magnetic metalorganic materials
2020. Johan Hellsvik (et al.). Physical Review Materials 4 (2)
ArticleRead more about Spin wave excitations of magnetic metalorganic materialsThe Organic Materials Database (OMDB) is an open database hosting about 22 000 electronic band structures, density of states, and other properties for stable and previously synthesized three-dimensional organic crystals. The web interface of the OMDB offers various search tools for the identification of novel functional materials such as band structure pattern matching and density of states similarity search. In this work, the OMDB is extended to include magnetic excitation properties. For inelastic neutron scattering, we focus on the dynamic structure factor S(q, omega) which contains information on the excitation modes of the material. We introduce a new dataset containing atomic magnetic moments and Heisenberg exchange parameters for which we calculate the spin wave spectra and dynamic structure factor with linear spin wave theory and atomistic spin dynamics. We thus develop the materials informatics tools to identify novel functional organic and metalorganic magnets.
-
Band Gap Prediction for Large Organic Crystal Structures with Machine Learning
2019. Bart Olsthoorn (et al.). Advanced Quantum Technologies 2 (7-8)
ArticleRead more about Band Gap Prediction for Large Organic Crystal Structures with Machine LearningMachine‐learning models are capable of capturing the structure–property relationship from a dataset of computationally demanding ab initio calculations. Over the past two years, the Organic Materials Database (OMDB) has hosted a growing number of calculated electronic properties of previously synthesized organic crystal structures. The complexity of the organic crystals contained within the OMDB, which have on average 82 atoms per unit cell, makes this database a challenging platform for machine learning applications. In this paper, the focus is on predicting the band gap which represents one of the basic properties of a crystalline material. With this aim, a consistent dataset of 12 500 crystal structures and their corresponding DFT band gap are released, freely available for download at https://omdb.mathub.io/dataset. An ensemble of two state‐of‐the‐art models reach a mean absolute error (MAE) of 0.388 eV, which corresponds to a percentage error of 13% for an average band gap of 3.05 eV. Finally, the trained models are employed to predict the band gap for 260 092 materials contained within the Crystallography Open Database (COD) and made available online so that the predictions can be obtained for any arbitrary crystal structure uploaded by a user.
-
Computational search for Dirac and Weyl nodes in f-electron antiperovskites
2019. Anna Pertsova (et al.). Physical Review B 99 (20)
ArticleRead more about Computational search for Dirac and Weyl nodes in f-electron antiperovskitesWe present the result of an ab initio search for new Dirac materials among inverse perovskites. Our investigation is focused on the less studied class of lanthanide antiperovskites containing heavy f-electron elements in the cation position. Some of the studied compounds have not yet been synthesized experimentally. Our computational approach is based on density functional theory calculations which account for spin-orbit interaction and strong correlations of the f-electron atoms. We find several promising candidates among lanthanide antiperovskites which host bulk Dirac states close to the Fermi level. Specifically, our calculations reveal massive three-dimensional Dirac states in materials of the class A(3)BO, where A=Sm, Eu, Gd, Yb, and B=Sn, Pb. In materials with finite magnetic moment, such as Eu3BO (B=Sn, Pb), the degeneracy of the Dirac nodes is lifted, leading to appearance of Weyl nodes.
-
Hund nodal line semimetals
2019. R. Matthias Geilhufe, Francisco Guinea, Vladimir Juričić. Physical Review B 99 (2)
ArticleRead more about Hund nodal line semimetalsWe propose a class of topological metals, which we dub Hund nodal line semimetals, arising from the strong Coulomb interaction encoded in the Hund's coupling between itinerant electrons and localized spins. We here consider a particular twisted spin configuration, which is realized in the double-exchange model which describes the manganite oxides. The resulting effective tetragonal lattice of electrons with hoppings tied to the local spin features an antiunitary nonsymmorphic symmetry that, in turn, together with another nonsymmorphic but unitary glide-mirror symmetry, protects crossings of a double pair of bands along a high-symmetry line on the Brillouin zone boundary. We also discuss the stability of Hund nodal line semimetals with respect to symmetry breaking arising from various perturbations of the twisted phase. Our results motivate further studies of other realizations of this state of matter, for instance, in different spin backgrounds, properties of its drumhead surface states, as well as its stability to disorder and interactions among the itinerant electrons.
-
Chemical-Strain Induced Tilted Dirac Nodes in (BEDT-TTF)(2)X-3 (X = I, Cl, Br, F) Based Charge-Transfer Salts
2018. R. Matthias Geilhufe, Benjamin Commeau, Gayanath W. Fernando. Physica Status Solidi. Rapid Research Letters 12 (11)
ArticleRead more about Chemical-Strain Induced Tilted Dirac Nodes in (BEDT-TTF)(2)X-3 (X = I, Cl, Br, F) Based Charge-Transfer SaltsThe identification of novel multifunctional Dirac materials has been an ongoing effort. In this connection quasi 2-dimensional (BEDT-TTF)-based charge transfer salts are widely discussed. Here, we report about the electronic structure of alpha-(BEDT-TTF)(2)I-3 and kappa-(BEDT-TTF)(2)I-3 under a hypothetical substitution of iodine with the halogens bromine, chlorine, and fluorine. The decreasing size of the anion layer corresponds to applying chemical strain which increases tremendously in the case of (BEDT-TTF)(2)F-3. We perform structural optimization and electronic structure calculations in the framework of density functional theory, incorporating, first, the recently developed strongly constrained and appropriately normed semilocal density functional SCAN, and, second, van der Waals corrections to the PBE exchange correlation functional by means of the dDsC dispersion correction method. In the case of alpha-(BEDT-TTF)(2)F-3, the formation of over-tilted Dirac-type-II nodes within the quasi two-dimensional Brillouin zone can be found. For kappa-(BEDT-TTF)(2)F-3, the recently reported topological transition within the electronic band structure cannot be revealed.
-
GTPack
2018. R. Matthias Geilhufe, Wolfram Hergert. Frontiers in Physics 6
ArticleRead more about GTPackWe present the Mathematica group theory package GTPack providing about 200 additional modules to the standard Mathematica language. The content ranges from basic group theory and representation theory to more applied methods like crystal field theory, tight-binding and plane-wave approaches capable for symmetry based studies in the fields of solid-state physics and photonics. GTPack is freely available via http://GTPack.org. The package is designed to be easily accessible by providing a complete Mathematica-style documentation, an optional input validation and an error strategy. We illustrate the basic framework of the package and show basic examples to present the functionality. Furthermore, we give a complete list of the implemented commands including references for algorithms within the Supplementary Material.
-
Materials Informatics for Dark Matter Detection
2018. R. Matthias Geilhufe (et al.). Physica Status Solidi. Rapid Research Letters 12 (11)
ArticleRead more about Materials Informatics for Dark Matter DetectionDark Matter particles are commonly assumed to be weakly interacting massive particles (WIMPs) with a mass in the GeV to TeV range. However, recent interest has shifted toward lighter WIMPs, which are more difficult to probe experimentally. A detection of sub-GeV WIMPs will require the use of small gap materials in sensors. Using recent estimates of the WIMP mass, we identify the relevant target space toward small gap materials (100 to 10 meV). Dirac Materials, a class of small- or zero-gap materials, emerge as natural candidates for sensors for Dark Matter detection. We propose the use of informatics tools to rapidly assay materials band structures to search for small gap semiconductors and semimetals, rather than focusing on a few preselected compounds. As a specific example of the proposed strategy, we use the organic materials database () to identify organic candidates for sensors: the narrow band gap semiconductors BNQ-TTF and DEBTTT with gaps of 40 and 38 meV, and the Dirac-line semimetal (BEDT-TTF)center dot Br which exhibits a tiny gap of approximate to 50 meV when spin-orbit coupling is included. We outline a novel and powerful approach to search for dark matter detection sensor materials by means of a rapid assay of materials using informatics tools.
-
Online search tool for graphical patterns in electronic band structures
2018. Stanislav S. Borysov (et al.). Npj Computational Materials 4
ArticleRead more about Online search tool for graphical patterns in electronic band structuresMany functional materials can be characterized by a specific pattern in their electronic band structure, for example, Dirac materials, characterized by a linear crossing of bands; topological insulators, characterized by a Mexican hat pattern or an effectively free electron gas, characterized by a parabolic dispersion. To find material realizations of these features, manual inspection of electronic band structures represents a relatively easy task for a small number of materials. However, the growing amount of data contained within modern electronic band structure databases makes this approach impracticable. To address this problem, we present an automatic graphical pattern search tool implemented for the electronic band structures contained within the Organic Materials Database. The tool is capable of finding user-specified graphical patterns in the collection of thousands of band structures from high-throughput calculations in the online regime. Using this tool, it only takes a few seconds to find an arbitrary graphical pattern within the ten electronic bands near the Fermi level for 26,739 organic crystals. The source code of the developed tool is freely available and can be adapted to any other electronic band structure database.
-
Reply to Comment on 'Instability of the topological surface state in Bi2Se3 upon deposition of gold'
2018. A. Polyakov (et al.). Physical Review B 98 (13)
ArticleRead more about Reply to Comment on 'Instability of the topological surface state in Bi2Se3 upon deposition of gold'In the Comment on our publication [Phys. Rev. B 95, 180202(R) (2017)], R. A. Gordon claims that our main conclusion is not valid, namely that gold atoms deposited in situ on the (0001) surface of single-crystalline Bi2Se3 reside in substitutional sites, i.e., replacing bismuth atoms within the topmost quintuple layer (QL). Based on x-ray absorption near-edge (XANES) spectra and a re-evaluation of extended x-ray absorption fine structure (EXAFS) data above the Au L-III edge, R. A. Gordon concludes that Au resides in a twofold environment as a result of an interface reaction leading to an Au2S-type local structure, in which gold adopts an Au(I) state and is linearly coordinated by selenium atoms. In this Reply, we will confirm the results published in the original paper and their interpretation that Au atoms reside in the substitutional site.
-
Symmetry analysis of odd- and even-frequency superconducting gap symmetries for time-reversal symmetric interactions
2018. R. Matthias Geilhufe, Alexander V. Balatsky. Physical Review B 97 (2)
ArticleRead more about Symmetry analysis of odd- and even-frequency superconducting gap symmetries for time-reversal symmetric interactionsOdd-frequency superconductivity describes a class of superconducting states where the superconducting gap is an odd function in relative time and Matsubara frequency. We present a group theoretical analysis based on the linearized gap equation in terms of Shubnikov groups of the second kind. By discussing systems with spin-orbit coupling and an interaction kernel which is symmetric under the reversal of relative time, we show that both even-and odd-frequency gaps are allowed to occur. Specific examples are discussed for the square lattice, the octahedral lattice, and the tetragonal lattice. For irreducible representations that are even under the reversal of relative time the common combinations of s- and d-wave spin singlet and p-wave spin triplet gaps are revealed, irreducible representations that are odd under reversal of relative time give rise to s- and d-wave spin triplet and p-wave spin singlet gaps. Furthermore, we discuss the construction of a generalized Ginzburg-Landau theory in terms of the associated irreducible representations. The result complements the established classification of superconducting states of matter.
-
Towards novel organic high-T-c superconductors
2018. R. Matthias Geilhufe (et al.). Physical Review Materials 2 (2)
ArticleRead more about Towards novel organic high-T-c superconductorsIdentifying novel functional materials with desired key properties is an important part of bridging the gap between fundamental research and technological advancement. In this context, high-throughput calculations combinedwith data-mining techniques highly accelerated this process in different areas of research during the past years. The strength of a data-driven approach for materials prediction lies in narrowing down the search space of thousands of materials to a subset of prospective candidates. Recently, the open-access organic materials database OMDBwas released providing electronic structure data for thousands of previously synthesized three-dimensional organic crystals. Based on the OMDB, we report about the implementation of a novel density of states similarity search tool which is capable of retrieving materials with similar density of states to a reference material. The tool is based on the approximate nearest neighbor algorithm as implemented in the ANNOY library and can be applied via the OMDB web interface. The approach presented here is wide ranging and can be applied to various problems where the density of states is responsible for certain key properties of a material. As the first application, we report about materials exhibiting electronic structure similarities to the aromatic hydrocarbon p-terphenyl which was recently discussed as a potential organic high-temperature superconductor exhibiting a transition temperature in the order of 120 K under strong potassium doping. Although the mechanism driving the remarkable transition temperature remains under debate, we argue that the density of states, reflecting the electronic structure of a material, might serve as a crucial ingredient for the observed high T-c. To provide candidates which might exhibit comparable properties, we present 15 purely organic materials with similar features to p-terphenyl within the electronic structure, which also tend to have structural similarities with p-terphenyl such as space group symmetries, chemical composition, and molecular structure. The experimental verification of these candidates might lead to a better understanding of the underlying mechanism in case similar superconducting properties are revealed.
-
Data Mining for Three-Dimensional Organic Dirac Materials
2017. R. Matthias Geilhufe (et al.). Scientific Reports 7
ArticleRead more about Data Mining for Three-Dimensional Organic Dirac MaterialsWe combined the group theory and data mining approach within the Organic Materials Database that leads to the prediction of stable Dirac-point nodes within the electronic band structure of three-dimensional organic crystals. We find a particular space group P2(1)2(1)2(1) (#19) that is conducive to the Dirac nodes formation. We prove that nodes are a consequence of the orthorhombic crystal structure. Within the electronic band structure, two different kinds of nodes can be distinguished: 8-fold degenerate Dirac nodes protected by the crystalline symmetry and 4-fold degenerate Dirac nodes protected by band topology. Mining the Organic Materials Database, we present band structure calculations and symmetry analysis for 6 previously synthesized organic materials. In all these materials, the Dirac nodes are well separated within the energy and located near the Fermi surface, which opens up a possibility for their direct experimental observation.
-
Instability of the topological surface state in Bi2Se3 upon deposition of gold
2017. A. Polyakov (et al.). Physical Review B 95 (18)
ArticleRead more about Instability of the topological surface state in Bi2Se3 upon deposition of goldMomentum-resolved photoemission spectroscopy indicates the instability of the Dirac surface state upon deposition of gold on the (0001) surface of the topological insulator Bi2Se3. Based on the structure model derived from extended x-ray absorption fine structure experiments showing that gold atoms substitute bismuth atoms, first-principles calculations provide evidence that a gap appears due to hybridization of the surface state with gold d states near the Fermi level. Our findings provide insights into the mechanisms affecting the stability of the surface state.
-
Organic materials database
2017. Stanislav S. Borysov, R. Matthias Geilhufe, Alexander V. Balatsky. PLOS ONE 12 (2)
ArticleRead more about Organic materials databaseWe present an organic materials database (OMDB) hosting thousands of Kohn-Sham electronic band structures, which is freely accessible online at http://omdb.diracmaterials.org. The OMDB focus lies on electronic structure, density of states and other properties for purely organic and organometallic compounds that are known to date. The electronic band structures are calculated using density functional theory for the crystal structures contained in the Crystallography Open Database. The OMDB web interface allows users to retrieve materials with specified target properties using non-trivial queries about their electronic structure. We illustrate the use of the OMDB and how it can become an organic part of search and prediction of novel functional materials via data mining techniques. As a specific example, we provide data mining results for metals and semiconductors, which are known to be rare in the class of organic materials.
-
Structural and electronic properties of alpha-(BEDT-TTF)(2)I-3, ss-(BEDT-TTF)(2)I-3, and kappa-(BEDT-TTF)(2)X-3 (X = I, F, Br, Cl) organic charge transfer salts
2017. Benjamin Commeau (et al.). Physical Review B 96 (12)
ArticleRead more about Structural and electronic properties of alpha-(BEDT-TTF)(2)I-3, ss-(BEDT-TTF)(2)I-3, and kappa-(BEDT-TTF)(2)X-3 (X = I, F, Br, Cl) organic charge transfer salts(BEDT-TFF)(2)I-3 charge transfer salts are reported to show superconductivity and pressure-induced quasi-twodimensional Dirac cones at the Fermi level. By performing state of the art ab initio calculations in the framework of density functional theory, we investigate the structural and electronic properties of the three structural phases alpha, beta, and kappa(.) We furthermore report about the irreducible representations of the corresponding electronic band structures, symmetry of their crystal structure, and the origin of band crossings. Additionally, we discuss the chemically induced strain in kappa-(BEDT-TTF)(2)I-3 achieved by replacing the iodine layer with other halogens: fluorine, bromine, and chlorine. In the case of kappa-(BEDT-TTF)(2)F-3, we identify topologically protected crossings within the band structure. These crossings are forced to occur due to the nonsymmorphic nature of the crystal. The calculated electronic structures presented here are added to the organic materials database (OMDB).
-
Three-dimensional organic Dirac-line materials due to nonsymmorphic symmetry
2017. R. Matthias Geilhufe (et al.). Physical Review B 95 (4)
ArticleRead more about Three-dimensional organic Dirac-line materials due to nonsymmorphic symmetryA datamining study of electronic Kohn-Sham band structures was performed to identify Dirac materials within the Organic Materials Database. Out of that, the three-dimensional organic crystal 5,6-bis(trifluoromethyl)-2-methoxy-1H-1,3-diazepine was found to host different Dirac-line nodes within the band structure. From a group theoretical analysis, it is possible to distinguish between Dirac-line nodes occurring due to twofold degenerate energy levels protected by the monoclinic crystalline symmetry and twofold degenerate accidental crossings protected by the topology of the electronic band structure. The obtained results can be generalized to all materials having the space group P2(1)/c (No. 14, C-2h(5)) by introducing three distinct topological classes.
Show all publications by Matthias Geilhufe at Stockholm University